La-Proteina by NVIDIA: a new path for protein generation at full-atom scale
Our take on La-Proteina: what feels genuinely new, what holds up, and why it matters.
Last month, NVIDIA open sourced La-Proteina, an atomistic protein generation model built on a preprint released in July, and it caught more than a few readers off guard. If your first thought was “Wait, NVIDIA does biology?”, you are not the only one. The reality is that they have been in life sciences for years. You may know BioNeMo (NVIDIA’s platform for generative biology with GPU-ready models and services for training and inference) and Clara (the broader life-sciences stack spanning imaging, genomics, and computational drug discovery workflows). In that context, this work is not an outlier; it continues an infrastructure-first effort to make protein modeling practical at scale. When NVIDIA releases a protein modeling method, it is usually engineered for production, with attention to throughput, memory, batching, and execution across multiple GPUs, rather than a purely academic prototype. That does not make a method “good by brand”, but it often aligns with real pipeline needs: containerized inference, reproducibility, and clean handoffs to docking and MD. Our focus here is strictly technical: what is new, what actually scales, and where it fits in a working workflow.
See the preprint here: https://arxiv.org/abs/2507.09466
A Clear Overview of the Model
Let us begin with a broad view of La-Proteina, so the logic is clear before we step into specifics. In essence, it is a model for protein generation at full-atom resolution. The idea is simple to state and careful in execution. Keep the C-alpha backbone explicit so that global geometry stays under direct control. Compress residue identity and side-chain atoms into a compact latent vector for each residue. Learn to generate the backbone and those vectors together with flow matching, then use a variational autoencoder (VAE) to recover all-atoms and the sequence. In practice, it draws a clear line between the global structure and the local chemistry, which makes the problem easier to scale and easier to reason about.
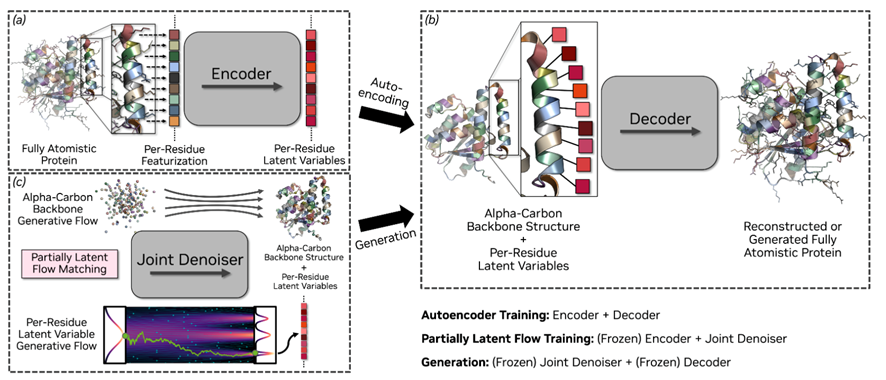
Before this work, most practical pipelines fell into two groups. The first designed only the backbone, for example with RFdiffusion, then added side-chains and sequence with other tools like rotamer packers or ProteinMPNN, and finally checked the result with AlphaFold or a short MD run. This can work, but it often drifts: a good fold can clash with later side-chains, motif rules are enforced mostly at the backbone level, and quality tends to drop as proteins get longer, so you end up redesigning. The second group tried full-atom co-design from the start, using recent all-atom generators, but working directly in atom space is heavy and fragile, so long sequences, large batches, and precise conditioning have been hard to do in practice.
La-Proteina tackles this by reframing the problem, not just changing the architecture. It keeps the backbone explicit and packs the per-residue chemistry into small, fixed-size codes. It learns to generate both together, then decodes back to all-atoms and the sequence. The payoff is practical: you can scaffold motifs at the atom level, including tip atom constraints that match chemical intent.
Results, Strengths, and Limits across Tasks
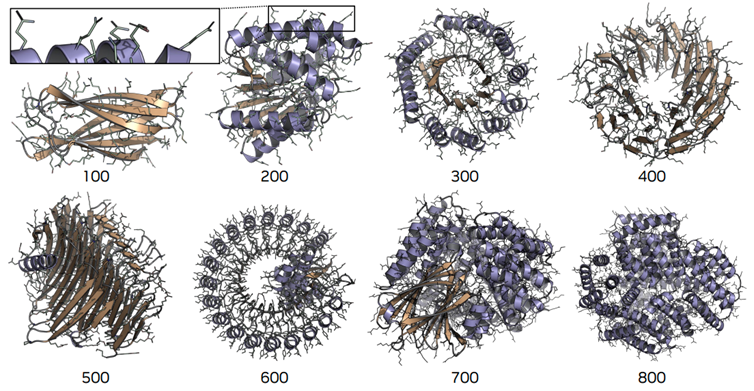
As interesting as the design is, the real test is what it enables in practice. The authors report that La-Proteina maintains quality in unconditional generation at lengths where other all-atom models struggle. In a length sweep, it produces co-designable proteins up to about 800 residues, while most comparison models either degrade in quality or run out of memory beyond roughly 500 residues. On standard structure quality measures, MolProbity (clashes, bond/angle outliers, Ramachandran) and rotamer statistics (favored side-chain conformations), La-Proteina's curves stay close to what you would expect from realistic structures as length increases, whereas several other models worsen. One comparison model required more than 140 GB of GPU memory to sample a single long sequence, so the authors capped it at 500 residues.
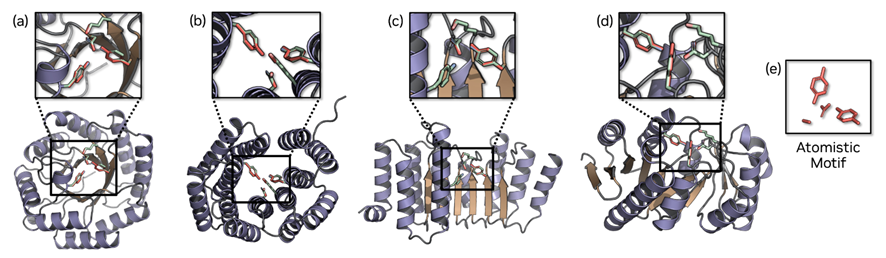
For motif scaffolding, they test all-atom and tip atom setups, each in indexed and unindexed forms, with strict success criteria, and report solving most of 26 tasks while the strongest baseline solves four; for motifs with three or more segments, unindexed sometimes performs better, likely because fixed indices over-constrain placement. On throughput, the model runs batched inference on an A100 with 80 GB, handling large batches and giving sub-second per-sample times for short sequences. This is helped by a light decoder, and in their comparison only La-Proteina and PLAID support true batching.
The work stays within single-chain proteins and trains mainly on curated sets from AlphaFold DB. To illustrate what the model can handle, the figures focus on enzyme active-site motifs, for example carbonic anhydrase and a retro aldolase, where the geometry of a few key atoms matters. The authors also note occasional small local quirks in side-chains (think an unusual rotamer or a ring flipped the wrong way), which they describe as rare. In our view, these are reasonable first release boundaries rather than red flags; they simply mean the present version does not yet cover complexes or ligand conditioned design and it does not include wet lab validation. Read the results as a strong in silico capability that is ready to be stress tested along those directions.
What You Can Run Now and What You Need
This release by NVIDIA includes La-Proteina with code and ready-to-use configs on GitHub, and the pretrained checkpoints on NVIDIA NGC. That is enough to generate proteins outright and to run motif scaffolding tasks where you fix a few atoms and let the model build the rest. The repo includes simple commands and example tasks, plus a small script to check basic quality, so you can try it without training from scratch.
Access the Github repo here: https://github.com/NVIDIA-Digital-Bio/la-proteina
Start with the GitHub README (or the NVIDIA Research project page) to set up the environment and download the weights from NGC. You will need an NVIDIA GPU; the authors benchmarked on an A100 (80 GB) and used the environment specified in the repo. More memory helps with long sequences and larger batches, but you can still test shorter sequences on smaller cards. The release focuses on single-chain proteins and atom level scaffolding; it does not cover multi-chain complexes or ligand conditioned design.
The bottom line for design pipelines
As described in NVIDIA's preprint, La-Proteina is positioned as a fast, upstream generator for single-chain proteins: the authors report full-atom proposals that scale to longer sequences and simple options to respect key motif geometry. In practice, that can make the "generate" step of a pipeline feel lighter, use it to draft many plausible starts, then let your usual filters decide what's worth keeping. It's not trying to replace downstream evaluation; it's a way to widen the top of the funnel.
If you choose to try it, a cautious, pipeline-friendly flow works well: generate a sizeable batch (add motif constraints only where they matter), refold with your standard tool and keep designs that come back consistently, cluster to avoid near-duplicates, run routine structure checks (clashes, geometry/rotamers) with a brief relax or short MD if needed, and then apply task-specific scoring, docking or interface metrics for binders, stability or packing for stand-alone folds. Two small tips from the authors' description: "tip-atom" constraints can help preserve active-site geometry, and lighter constraints on multi-segment motifs often give the scaffold room to settle. Batching is highlighted as a practical way to get throughput.
All of the above reflects what the authors report in the preprint; as with any new model, the picture will sharpen as more groups try it in different settings and, where relevant, in the lab. For now it targets single-chain design, and features like complexes or explicit ligand conditioning look like natural next steps rather than the target of this version. Even so, it's a meaningful step toward easing a real bottleneck: fast, motif-aware all-atom generation. If the reported scaling and scaffolding gains hold up, La-Proteina could become a very practical upstream generator in design workflows. Are you as excited as we are to try it?
Key Concepts
• Flow matching: Learn a continuous velocity field that transports simple noise into the data distribution. For La-Proteina this moves samples toward valid C-alpha coordinates and per-residue codes smoothly, which gives stable training and fast sampling.
• Joint generation: Model backbone geometry and residue-level codes together. The fold can inform local choices and local choices can inform the fold during sampling, which avoids the common mismatch of a good backbone with side-chains that do not fit.
• Fixed dimension latent per-residue: Each residue carries a constant size vector that summarizes identity and side-chain detail. Constant shapes are easy to batch on GPUs and scale to long sequences, while the decoder restores full-atoms later.
• Variational autoencoder (decoder): During training, full-atoms and sequence are encoded into the per-residue codes. At inference, the decoder reconstructs all-atoms and the amino acid sequence from those codes, so the generator works in a simpler space.
• Conditioning and scaffolding: You can fix a motif and ask the model to build around it. All-atom fixes the entire motif; tip-atom fixes only the functional atoms past the last rotatable bond. Indexed provides residue indices; unindexed provides only positions.
• Two clocks for sampling: Backbone and residue codes follow different time schedules during generation. Let the global fold settle first and let local details follow, which improves consistency for long chains.
• Co-design at full-atom resolution: Sequence and full-atom structure are decided in one process. This keeps rotamers, contacts, and hydrogen bonds in a realistic regime, which reduces cleanup in docking and short molecular dynamics.
• MolProbity: A bundle of clash, bond, angle, and Ramachandran scores. Lower clash and fewer outliers mean geometry that is closer to real structures.
• Rotamer statistics: How often side-chains fall into favored conformations seen in experimental data. Closer to observed distributions is better and helps downstream physics.
• Length and scaling: Check whether those metrics stay stable as sequence length grows. If quality holds at several hundred residues, the method is more likely to survive real workloads.
References
Geffner, T.; Didi, K.; Cao, Z.; Reidenbach, D.; Zhang, Z.; Dallago, C.; Kucukbenli, E.; Kreis, K.; Vahdat, A. La-Proteina: Atomistic protein generation via partially latent flow matching. arXiv 2025, arXiv:2507.09466.
NVIDIA Research. La-Proteina. https://research.nvidia.com/labs/genair/la-proteina/ (accessed 2025-09-13).
NVIDIA Digital Biology. la-proteina; GitHub. https://github.com/NVIDIA-Digital-Bio/la-proteina (accessed 2025-09-13).
Tags:
Contents
About the author

Iker Zapirain
Co-founder & CEO
Co-founder and CEO of Nomosis, leading the vision for AI-driven biological research and computational drug discovery.
Related articles
Molecular Dynamics Simulation
5 min read
AI in Biological Research
8 min read
Protein Design with AI
12 min read