Trajectory Viewer: Advanced MD Visualization & Analysis
Explore and analyze molecular dynamics trajectories with powerful visualization tools, comprehensive analysis capabilities, and interactive measurement features. Navigate through time and space to understand molecular behavior like never before.
Representations
Flexible visualization modes to explore your molecular dynamics trajectories from every angle
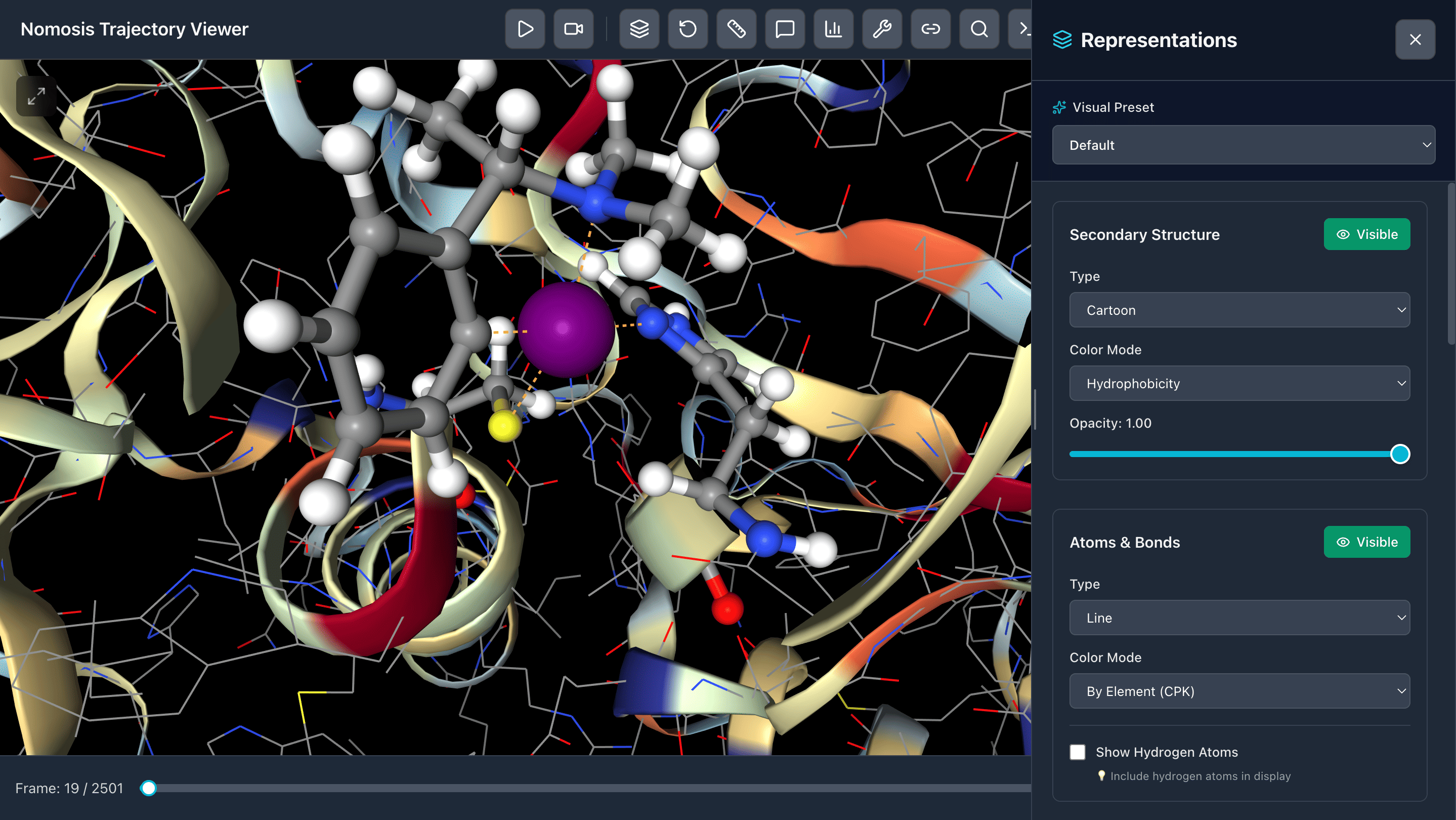
Change Representation Modes
Switch between cartoon, surface, sticks, spacefill, and custom representations in real-time to highlight different aspects of your molecular system.
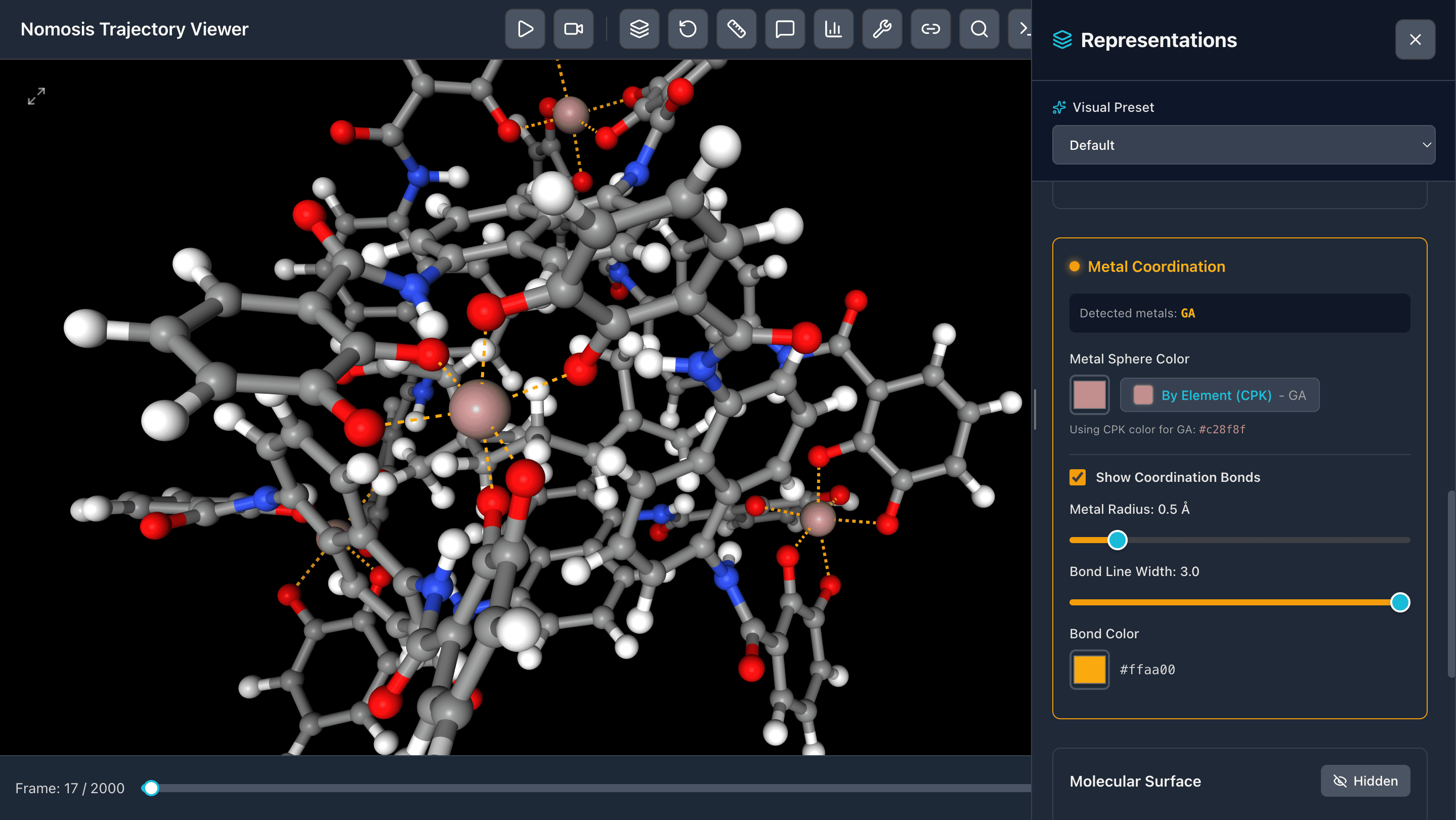
Show Metallic Bonds
Visualize coordination bonds and metallic interactions with customizable bond styles and colors for transition metals and coordination complexes.
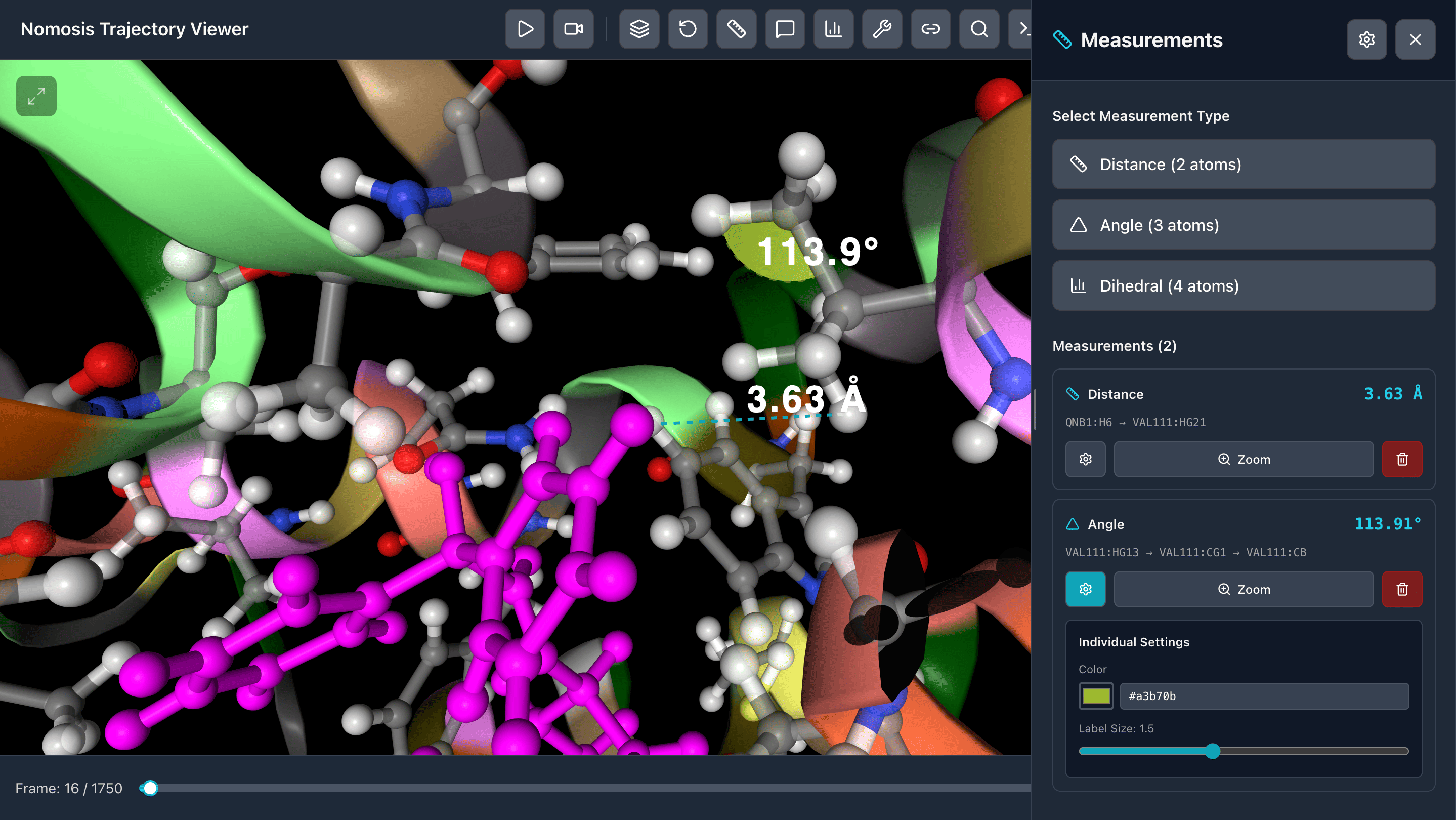
Measurements
Interactive measurement tools to analyze distances, angles, dihedrals, and other geometric parameters throughout your trajectory.
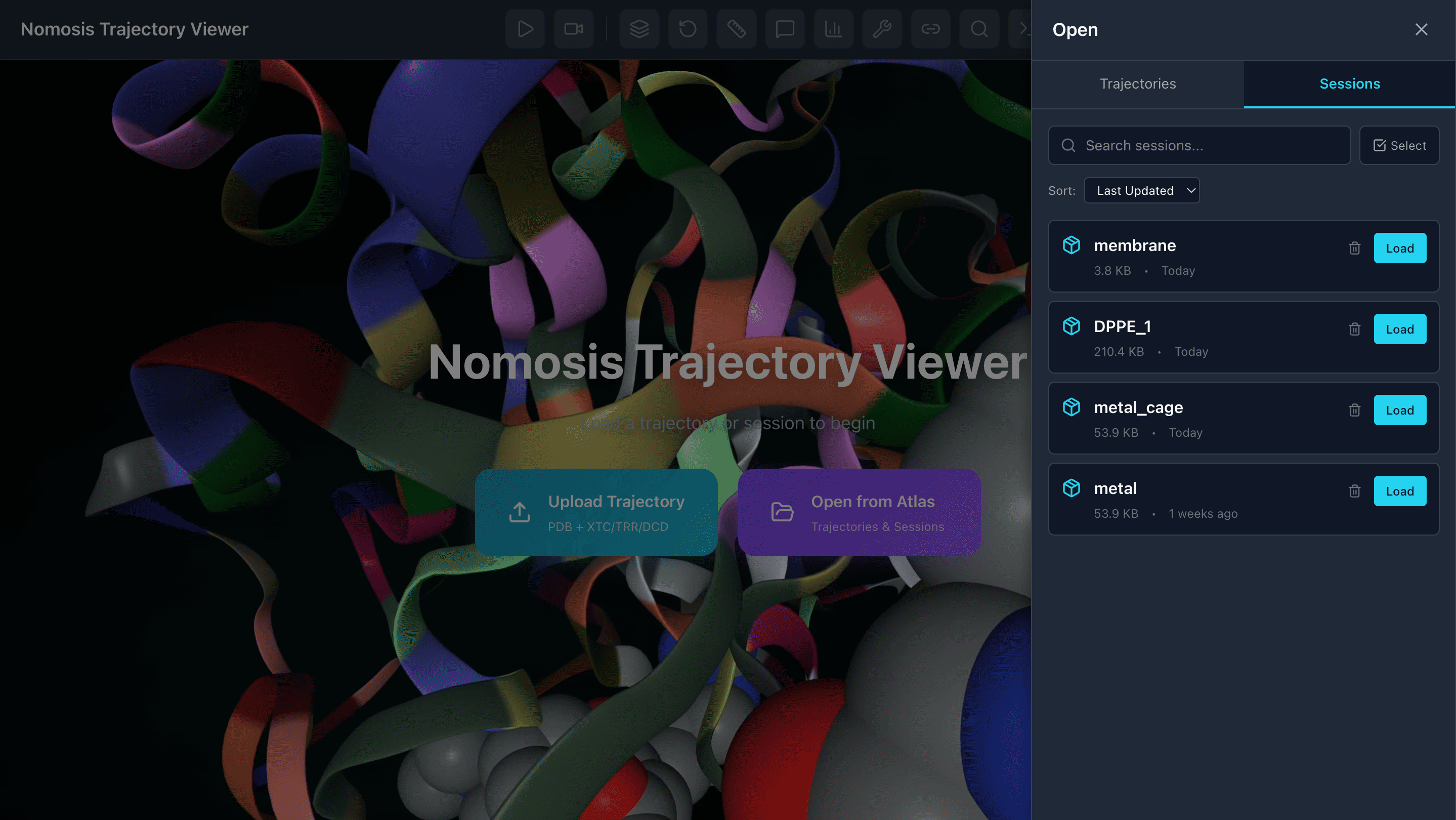
Load and Save Sessions
Save sessions with all your representations, calculations, and analyses to revisit them later. Load previous sessions to continue your work seamlessly.
Analysis
Comprehensive trajectory analysis tools to extract meaningful insights from your molecular dynamics simulations
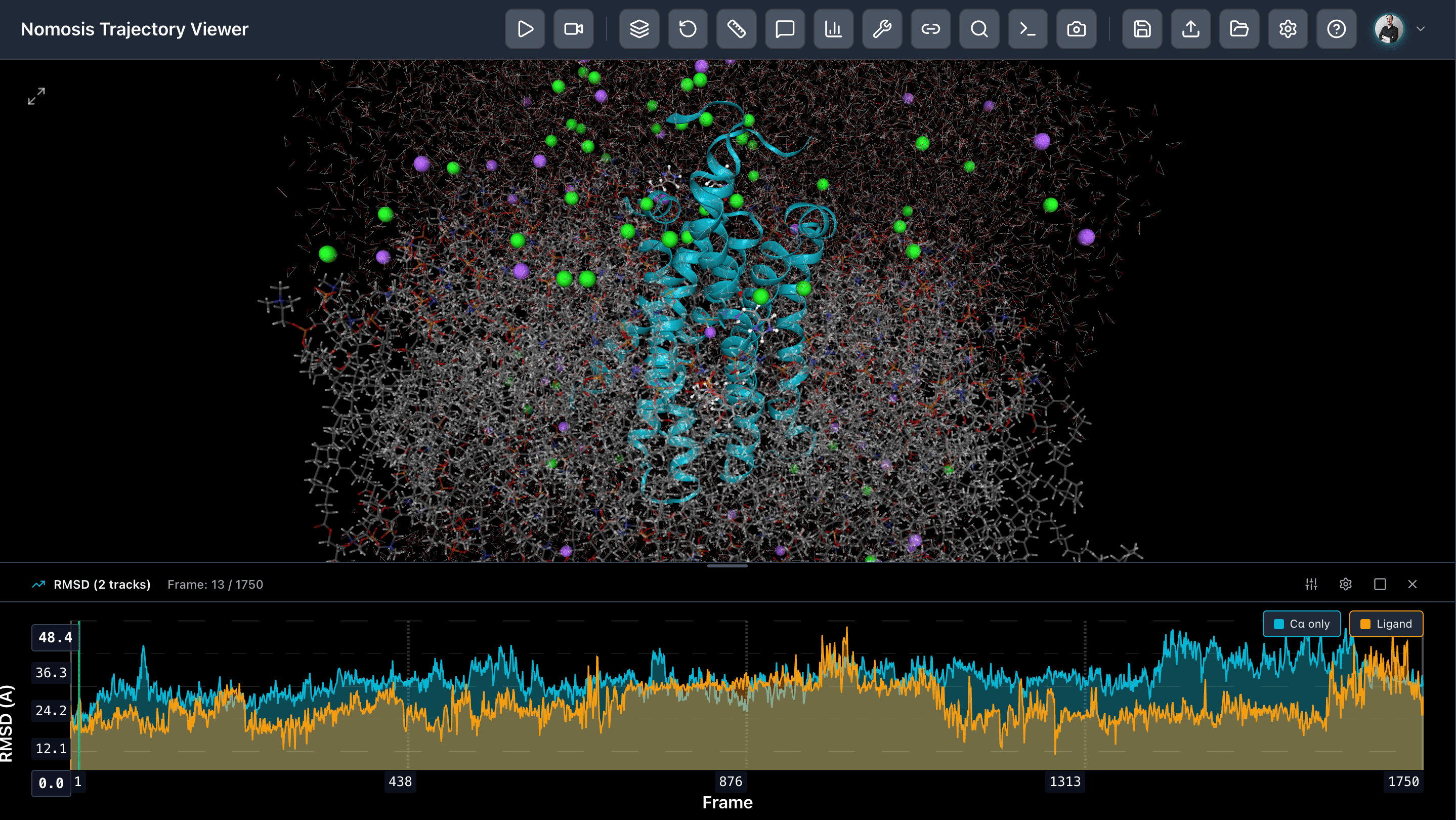
RMSD Analysis
Calculate and visualize Root Mean Square Deviation to track structural changes over time and identify conformational transitions.
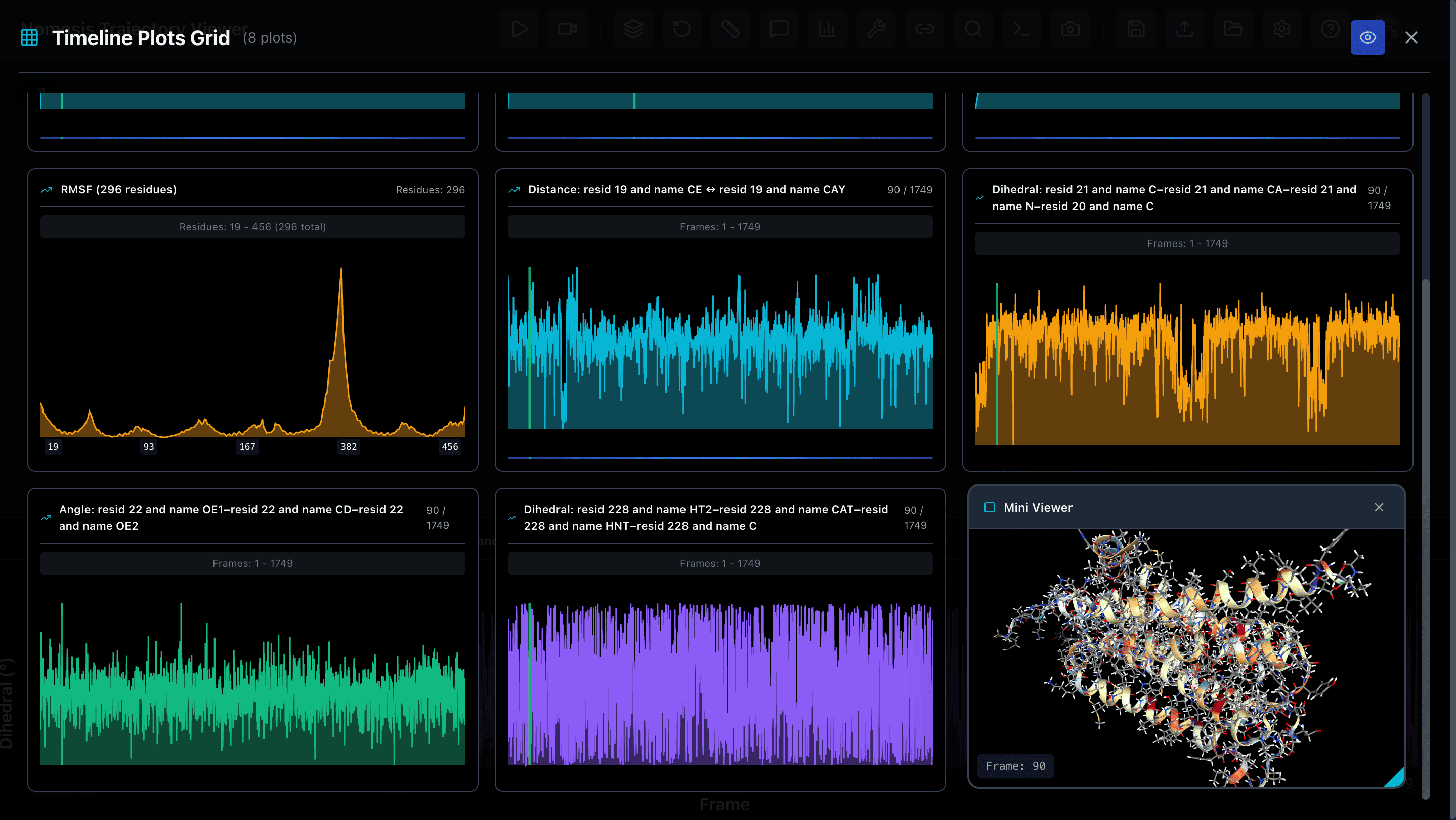
Multiplot Analysis
Visualize multiple analysis plots simultaneously with a mini trajectory viewer that keeps your trajectory visible at all times for contextual understanding.
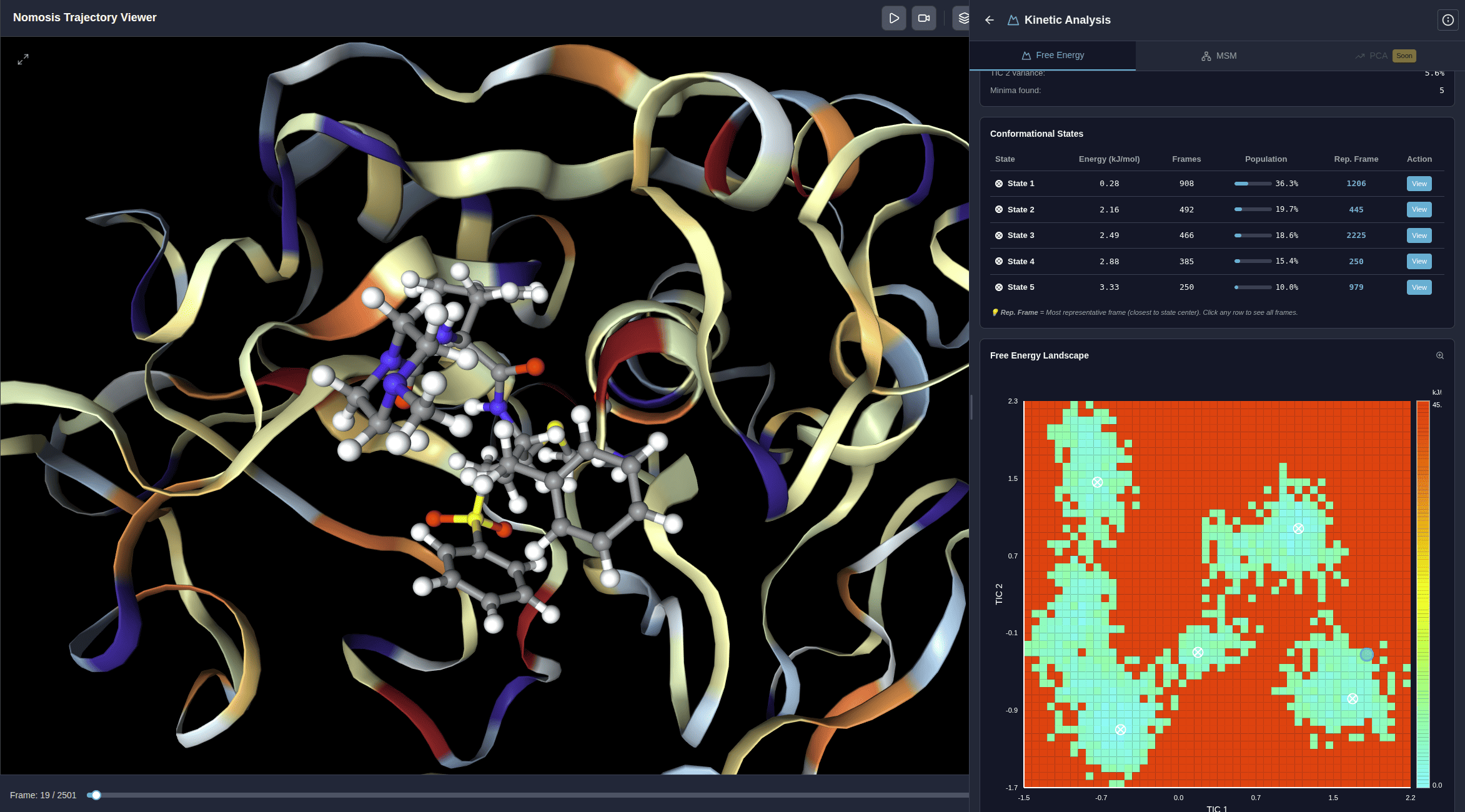
Microstate Analysis
Identify and cluster conformational microstates to understand the energy landscape and state transitions in your trajectory.
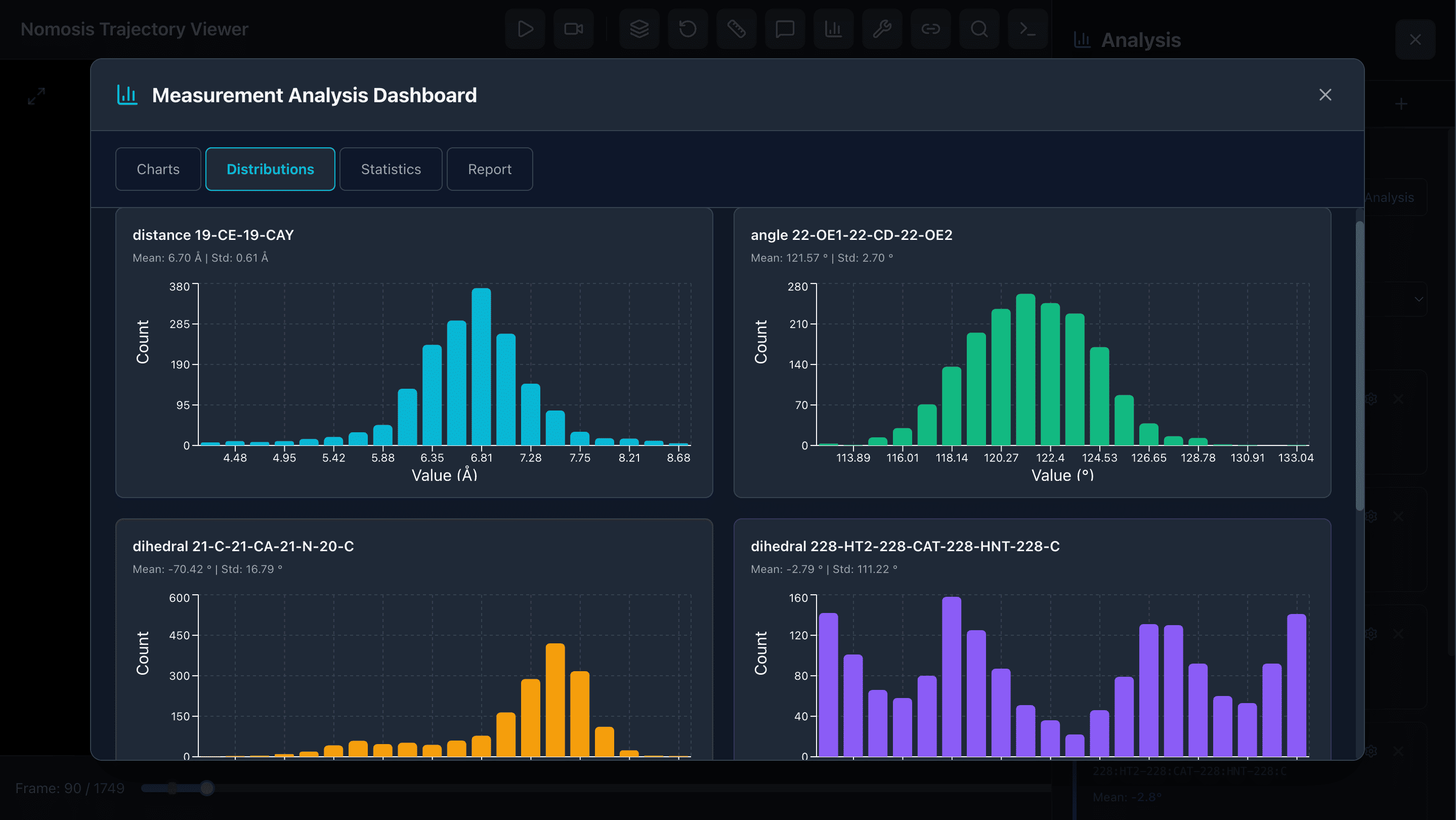
Advanced Metrics
Compute radius of gyration, solvent accessible surface area, hydrogen bond analysis, and other structural metrics over time.
Tools
Powerful measurement and analysis tools to extract quantitative insights from your trajectories
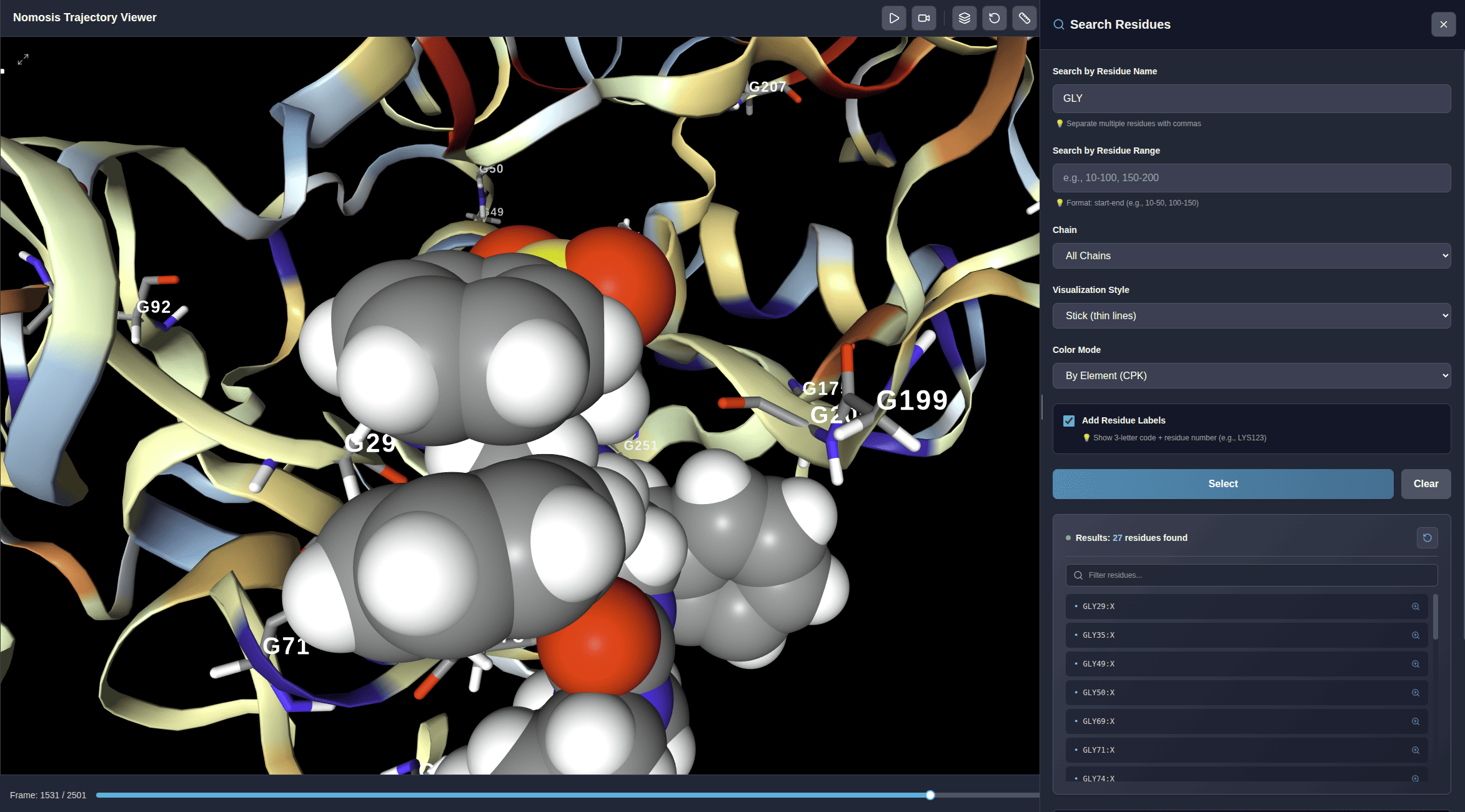
Search Residues
Search residues by number, name, and chain. Add labels and view results in a table. Click to zoom and highlight specific residues in your trajectory.
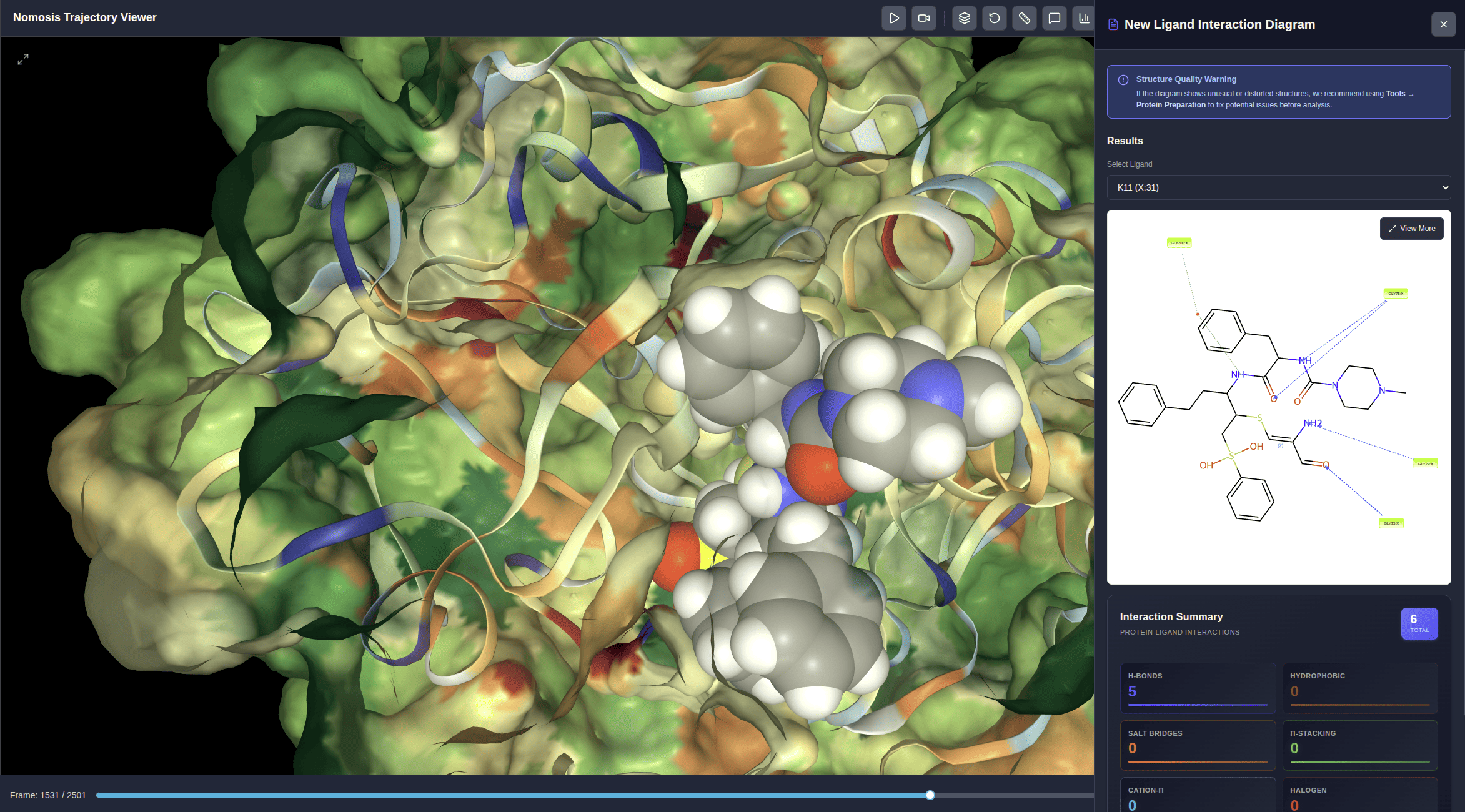
Ligand Interaction Diagram
Interactive 2D diagram showing which ligand atoms interact with which amino acids and the interaction types: hydrogen bonds, hydrophobic, π-π stacking, π-cation, water bridges, and more.
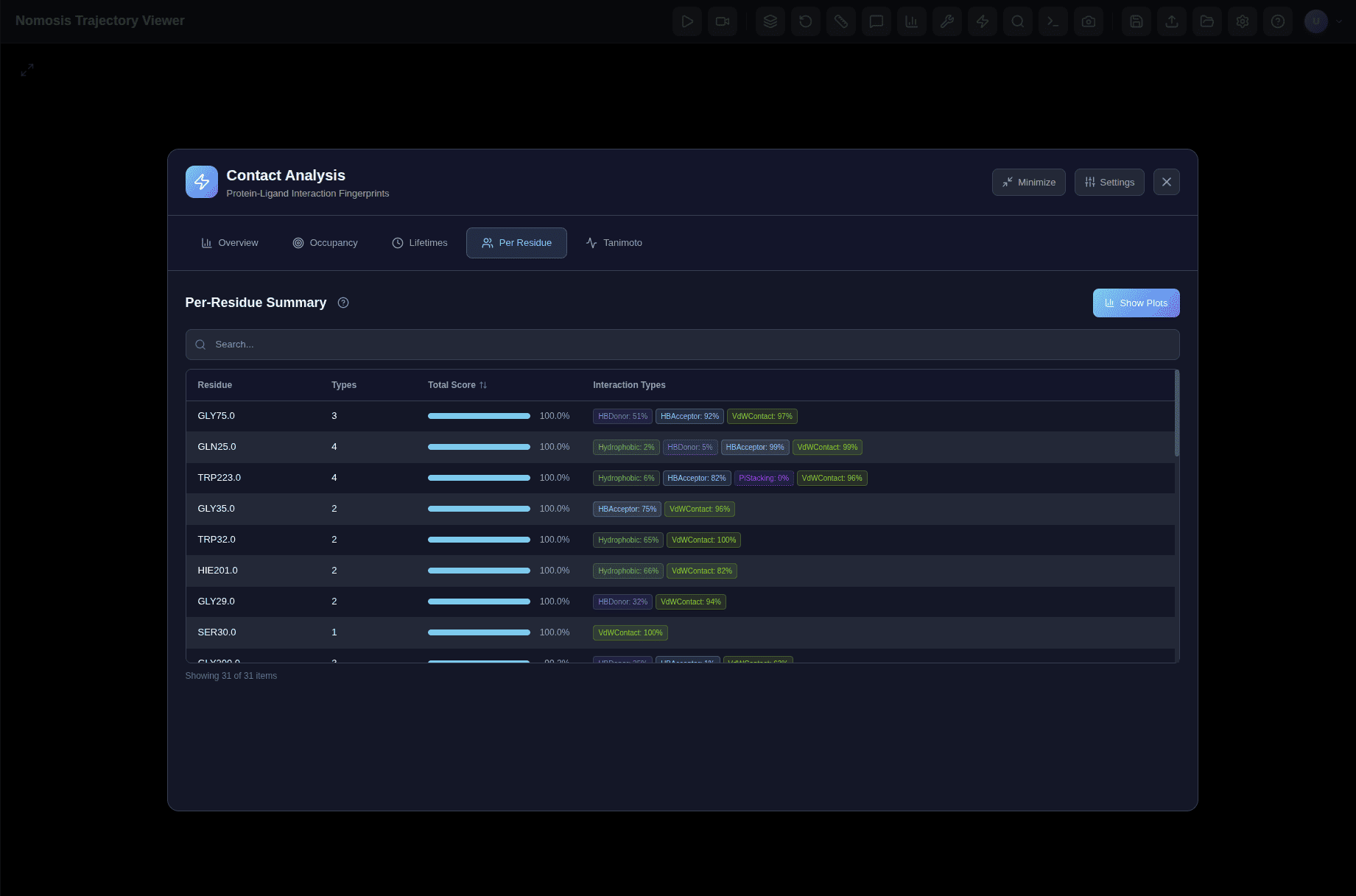
Ligand-Protein Interaction Analysis
Analyze binding interactions, identify contact residues, calculate binding energies, and track interaction patterns throughout the simulation.
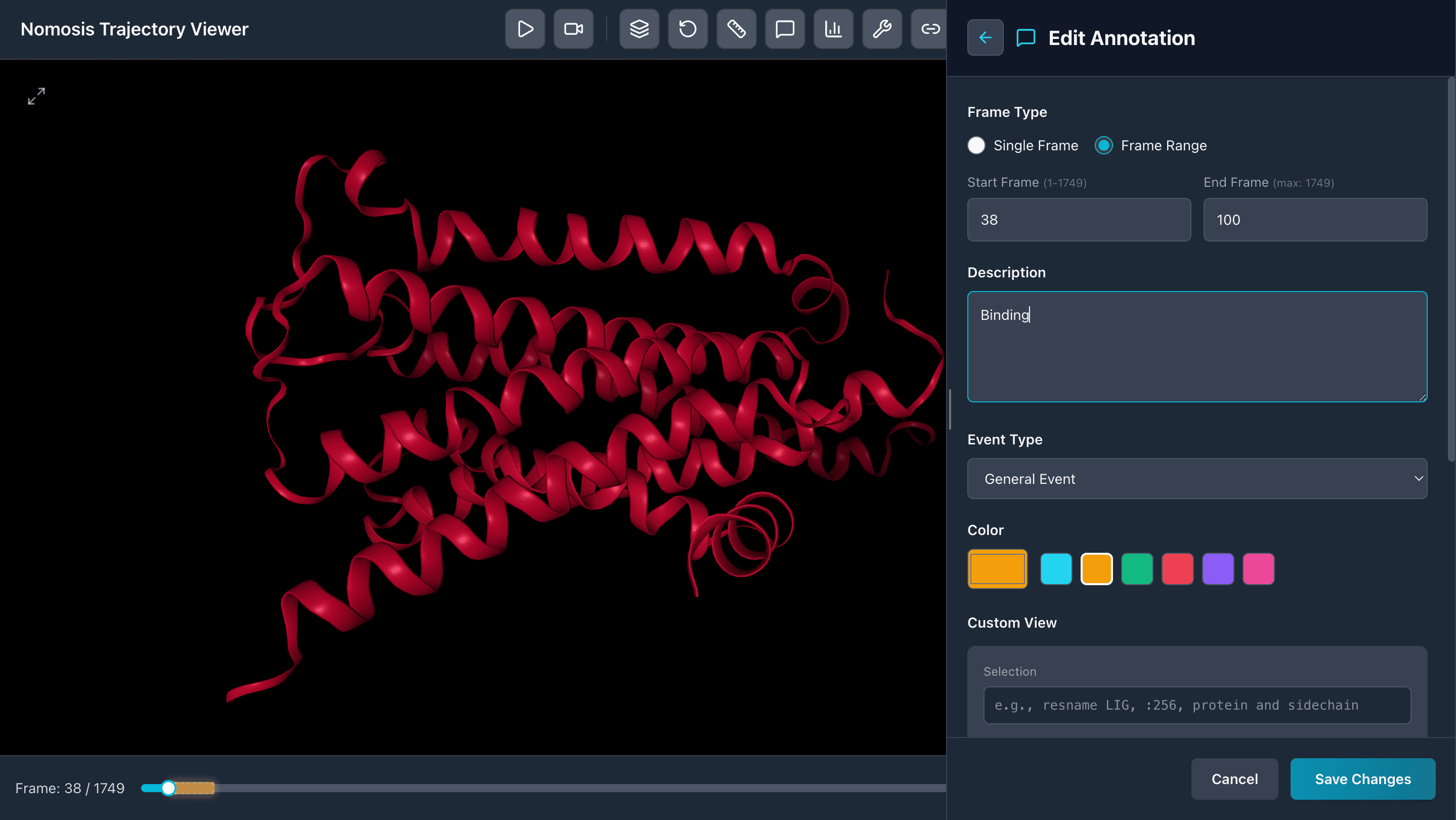
Frame Annotations
Create annotations for specific frames or frame ranges to mark important events, conformational changes, or analysis milestones in your trajectory.
Immersive Mode
Step into your molecular world with full-screen immersive visualization. Experience your trajectories in a distraction-free environment optimized for detailed analysis and exploration.
- Full-screen trajectory playback
- Enhanced rendering quality and performance
- Immersive navigation controls
- Perfect for presentations and demos
Ready to explore your trajectories?
Start visualizing and analyzing your molecular dynamics simulations with powerful tools designed for researchers.
Cookie Preferences
We use cookies to enhance your experience
We use cookies to analyze website traffic, personalize content, and improve your experience. You can customize your preferences or accept all cookies.